
Moderna a breveté la séquence du gène du SRAS-CoV2 « fabriqué par l’homme » en 2018
« CTCCTCGGCGGGCACGTAG » est le pistolet fumant du Covid-19 qui fera la lumière sur cette pandémie.
Le génome du SRAS-CoV-2 contient une séquence inhabituelle de 19 nucléotides que l’on ne trouve nulle part ailleurs dans la nature – à part dans les brevets Moderna soumis en 2018.
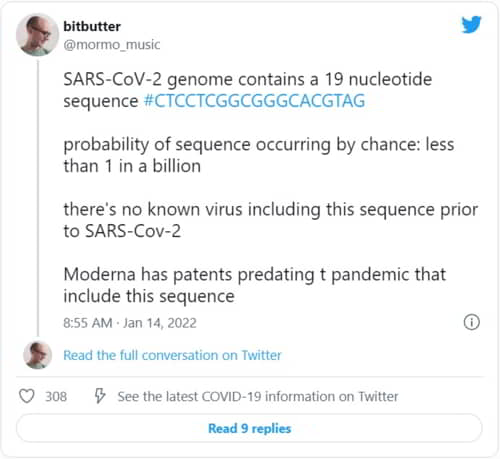
SARS-CoV-2 genome contains a 19 nucleotide sequence #CTCCTCGGCGGGCACGTAG
probability of sequence occurring by chance: less than 1 in a billion
there’s no known virus including this sequence prior to SARS-Cov-2
Moderna has patents predating t pandemic that include this sequence
— bitbutter (@mormo_music) January 14, 2022
Selon les recherches approfondies menées par le Dr Ah Kahn Syed, Moderna a breveté la séquence unique trouvée dans le coronavirus deux ans avant le début de la pandémie, ce qui renforce la théorie selon laquelle le Covid-19 est un virus créé par l’homme.
Le Dr Syed écrit : Cela fait longtemps que je veux écrire cet article. Eh bien, au moins depuis que Prashant Pradhan (un merveilleux, honnête et courageux scientifique en génomique) a évoqué la possibilité, en février 2020, que le virus SARS-Cov2 ait été fabriqué par l’homme. Et nous avons vu de nombreuses pièces confirmant que le virus a été fabriqué dans un laboratoire, l’une des meilleures ici sur zenodo et avec sa propre vidéo ici. À l’heure où j’écris ces lignes, ces liens sont toujours en place, ce qui, après 12 mois, est une bonne chose pour tout article qui ose remettre en question le radotage propagé par notre chère « presse libre [sponsorisée par l’industrie pharmaceutique] ».
Quoi qu’il en soit, BLAST est le dépôt du NCBI/NIH (alias le gouvernement américain) pour les séquences génomiques et protéomiques, entre autres choses. C’est là que tous les généticiens du monde entier déposent leurs séquences lorsqu’ils font une découverte. Sa principale fonction est de permettre la comparaison des séquences de gènes et la découverte de séquences qui correspondent à une séquence que vous avez peut-être rencontrée dans votre expérience. Qu’est-ce qu’une séquence de gènes ? C’est simple. C’est une ligne de code, composée de n’importe quelle combinaison de 4 lettres dans une séquence. Vous vous souvenez du film GATTACA ? Si vous ne l’avez pas encore regardé, vous devriez, car il s’agit d’un autre film dystopique qui est maintenant trop proche de chez vous.
Le titre du film est basé sur les 4 bases nucléotidiques (G, A, T, C) qui constituent le code génétique de l’ADN de chaque être humain. Il y en a environ 3 milliards dans chaque cellule, ce qui donne un code unique – ce qui fait de vous un individu unique ! Le code s’apparie de telle sorte que G-C et A-T se combinent toujours pour former la double hélice que vous voyez sur l’image, par exemple GATTACA serait apparié à CTAATGT (le complément). Le code est lu dans un sens spécifique, de sorte que GATTACA sur un brin serait TGTAATC sur l’autre (le complément inverse). L’un des avantages de BLAST est qu’il ne se soucie pas de la version que vous lui donnez, il vous indiquera toujours le bon gène.
Une autre chose à noter à ce stade concerne les probabilités. Vous l’avez fait à l’école en lançant une pièce de monnaie (où le code est H pour pile ou T pour face). Quelle serait la probabilité de HHHHH (1 sur 2^4 = 1/16). Il en va de même pour TTTT. Il en va de même pour THTH, ou toute autre séquence spécifique de tirages à pile ou face. Essayez vous-même si vous ne me croyez pas (prédisez d’abord la séquence et voyez ensuite combien de fois vous devez l’exécuter). Le code génétique est essentiellement une « pièce de monnaie à quatre faces ». Ainsi, pour toute exécution d’une séquence spécifique (par exemple GATC), la probabilité d’obtenir cette séquence EXACTE est de 1 sur 4^4, ou pour tout nombre n de nucléotides (nt ou bases), la probabilité est de 1 sur 4^n (ceci est simplifié car dans certaines situations, la probabilité que la base suivante soit X dépend des bases environnantes).
BLAST a deux sections – nucléotide (BLASTn) et protéine (BLASTp). BLASTp traite les séquences d’acides aminés, de la même manière que les séquences de nucléotides. Mais il y a une grande différence car il y a 20 acides aminés (au lieu de 4 nucléotides) et par conséquent, même les séquences courtes (par exemple QTNS = Glu-Thr-Asn-Ser) auraient une probabilité d’environ 1 sur 20^4 (simplifié), soit 1 sur 160 000. La probabilité qu’une séquence spécifique de 5 acides aminés apparaisse par hasard sur la même base s’élève à 1 sur 3,2 millions !
Appuyons donc sur le bouton Protein BLAST et c’est parti… et voici l’écran que vous obtiendrez, que je vais vous faire parcourir.
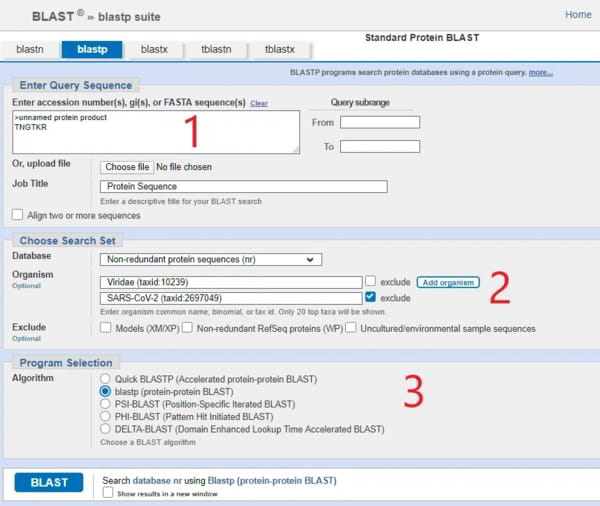
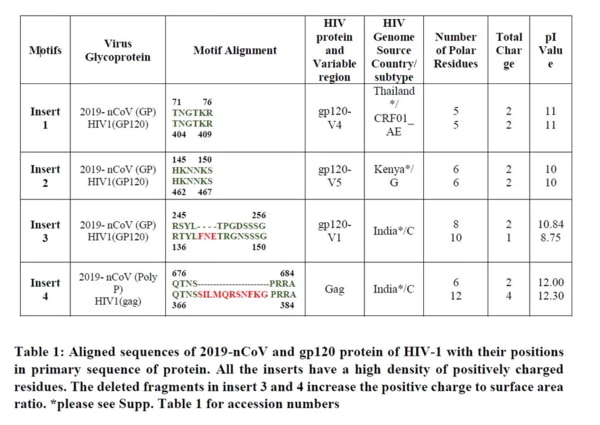
Dans [1], vous devez saisir la séquence d’acides aminés qui vous intéresse (BLAST ajoute automatiquement « >produit protéique sans nom »). Heureusement, vous n’avez pas besoin de chercher beaucoup car nous allons nous concentrer sur seulement 4 séquences dans le génome/protéome viral du SRAS-CoV-2 et celles-ci sont présentées dans le merveilleux article de Prashant Pradhan « Uncanny similarity of unique inserts in the 2019-nCoV spike protein to HIV-1 gp120 and Gag« publié le 31 janvier 2020 quelques jours après la publication de la séquence du génome.
La partie dont vous avez besoin se trouve dans le tableau 1 que je publie ici et vous verrez que j’ai affiché la séquence de 6 acides aminés TNGTKR dans la case [1] marquée en rouge sur l’écran BLASTp.
Pour l’étape [2] de l’écran de saisie de BLASTp, vous devez ajouter quelques filtres. Le premier filtre consiste à restreindre la recherche aux « viridae (virus) » (ou vous pouvez simplement entrer 10239 qui est l’ID taxonomique). La raison en est qu’il existe des milliards d’espèces sur la planète et que BLASTp les recherchera toutes, mais que vous voulez seulement savoir de quel virus provient ce motif. Il n’est pas vraiment intéressant de savoir si le motif est trouvé dans un calmar, bien qu’il soit possible qu’un calmar ait également participé à l’action avec la fameuse chauve-souris et le pangolin, dont parlent des gens comme Peter Daszak et Dominic Dwyer, ce qui en fait une ménagerie zoonotique, mais restons dans la réalité.
La deuxième condition est d’exclure toutes les références au SRAS-CoV-2 qui se sont maintenant accumulées dans la base de données, car elles vont toutes apparaître (des milliers) et cela ne nous intéresse pas.
Une fois que vous avez saisi ces données, appuyez sur le bouton BLAST et qu’obtenez-vous ? Vous obtiendrez une liste de candidats qui ont une homologie proche de cette séquence. Comme il s’agit d’une séquence très courte, l’homologie (ressemblance) devrait être de 100 %. Le haut de la page est un résumé de ce que vous avez demandé et le reste de la page est une liste des séquences correspondantes. Ce que vous verrez immédiatement, c’est qu’en haut de la liste se trouvent deux virus synthétiques qui sont une chimère du SRAS-Cov-2 et un autre virus, apparu au cours des deux dernières années grâce à des laboratoires qui fabriquent d’autres virus (parce que nous n’en avons pas assez). Le suivant dans la liste est un tas de références au VIH-1.
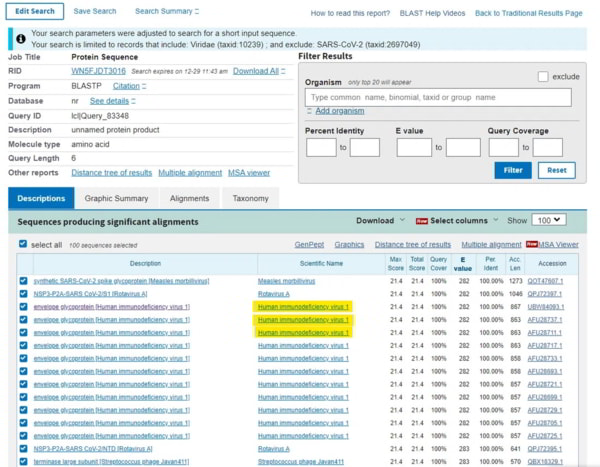
Vous pouvez cliquer sur n’importe lequel d’entre eux et vous serez amené à l’écran d’alignement où les alignements entre le sujet (votre TNGTKR) et la requête (tous les virus) sont affichés, et vous verrez en descendant la page que les alignements ne tiennent que pour le VIH-1 jusqu’à ce que vous commenciez à obtenir des protéines synthétiques et hypothétiques, jusqu’au prochain vrai virus dans la liste qui est le VIH-2….
OK, mais un résultat comme celui-ci pourrait être une coïncidence. Dans la liste, vous verrez la « valeur E » qui est un indicateur de la probabilité de trouver des correspondances comme celle-ci et qui doit être aussi proche de zéro que possible. Ici, elle est de 282, ce qui ne fait que refléter la probabilité de trouver des correspondances sur une séquence courte.
Donc, voilà le problème. Soit ces séquences vont par hasard correspondre à tout un tas d’autres virus (parce que la valeur E est élevée et que nous devons donc nous attendre à de nombreuses correspondances), soit il s’agit de séquences vraiment inhabituelles qui ont spécifiquement, préférentiellement ou uniquement correspondu au VIH-1. Comment aborder cette question ? Eh bien, passons à la séquence suivante – HKNNKS, qui est une autre séquence courte. Pour mettre de l’ordre dans l’écran, nous pouvons définir un filtre pour les séquences courtes afin de nous assurer que 100% de la séquence correspond (en haut à droite). Maintenant, souvenez-vous que si la correspondance avec le VIH-1 était aléatoire, nous ne devrions pas vraiment voir de correspondance sur cette liste, car elle devrait être éliminée par toutes les autres centaines de virus qui devraient correspondre de manière préférentielle. Oups…
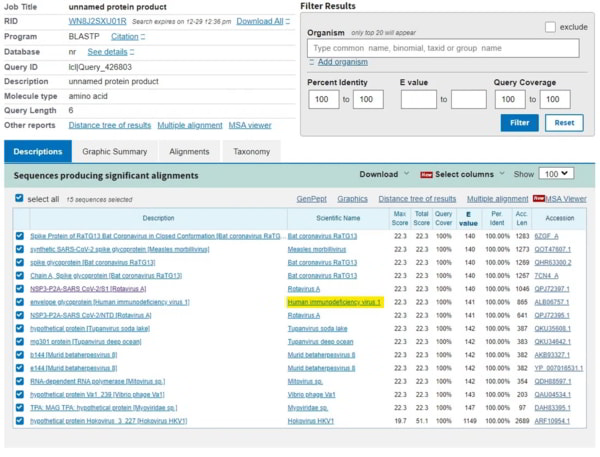
Notez les autres correspondances – Bat RaTG13 qui n’est apparu dans cette base de données qu’après que les gens aient commencé à s’interroger sur l’origine du coronavirus, et qui est probablement une séquence synthétique, et les mêmes virus synthétiques qui sont apparus après l’épidémie. Donc, le VIH-1 est la SEULE correspondance pour ces deux séquences.
Passons à la séquence 3. C’est une séquence plus longue. RSYLTPGDSSSG. Je me demande quel virus (ou quel ensemble de virus aléatoires, car c’est une séquence aléatoire, n’oubliez pas) correspondra à ….. Oh, regardez…
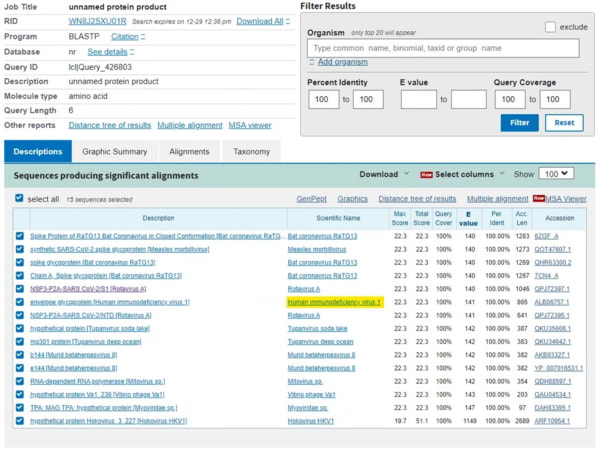
Dans cette recherche, j’ai donc limité la couverture de la requête à 100 % pour éliminer le bruit et les protéines hypothétiques. Il ne nous reste que les virus synthétiques de post-covid et RaTG13 (également post-covid). Le seul virus restant dans cette liste est, vous l’avez deviné, le VIH-1. Quelles sont les chances que le VIH-1 apparaisse dans les 3 recherches ?
Et pour compléter le quadruple tour, nous avons besoin de la dernière séquence d’insertion identifiée dans l’article de Pradhan, à savoir QTNS–PRRA. C’est une séquence très intéressante sur laquelle nous reviendrons plus tard, car c’est le site de clivage de la furine. C’est intéressant parce que les coronavirus bêta comme celui-ci n’ont pas de site de clivage de la furine, c’est le seul. Ce site ne peut sûrement pas provenir du VIH-1 ? Eh bien, il ne provient pas de la protéine GP120 comme les trois autres séquences, il est complètement différent et se trouve à un autre endroit du virus, ce que je vous montrerai bientôt, mais pour l’instant, exécutons le BLASTp.
Cette fois-ci, c’est un peu plus compliqué parce qu’un grand nombre de protéines hypothétiques et synthétiques ont été ajoutées depuis la publication de SARS-CoV-2 (j’aurais dû écrire cet article l’année dernière). Mais le VIH-1 fait à nouveau son apparition dans la liste et, cette fois, je ne montrerai que les alignements entre la protéine gag et le coronavirus – dans ce cas, il y a une délétion dans la protéine du VIH-1.
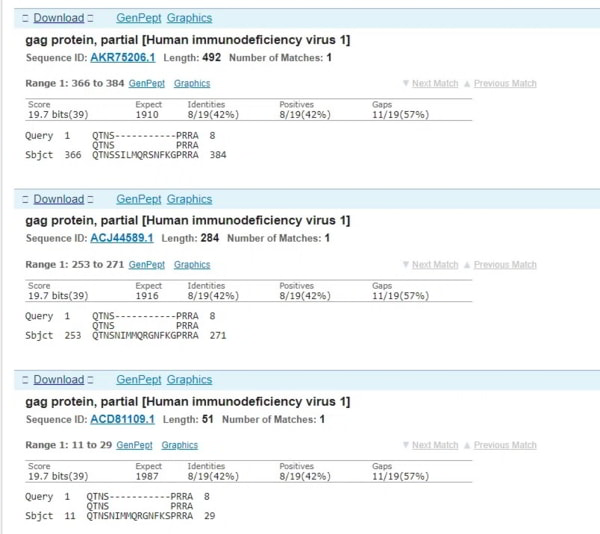
Nous avons donc 4 correspondances avec des séquences de VIH sans qu’aucun autre virus* n’apparaisse dans les 4 listes de correspondances (*sauf les virus synthétiques créés après l’événement). Quelles sont les chances de cela – proches de zéro.
Mais regardez ça. Il ne s’agissait pas simplement de séquences aléatoires du VIH. Dans son article, Pradhan est allé plus loin et a recréé la structure du virus avec l’emplacement des quatre insertions. Et voilà que ces insertions « aléatoires », toutes issues du VIH, se trouvent toutes sur des sites de liaison du coronavirus. Quelles sont les chances ?
Maintenant, il est possible que vous ne soyez pas convaincu. Malgré le fait que le seul virus à apparaître dans les listes de correspondance pour les quatre inserts, parmi les centaines de milliers de virus existants, se trouve être le VIH-1. Et le VIH-1 ne devrait avoir aucune chance réelle de former des virus recombinants avec les coronavirus de chauve-souris dans la nature, et aucune chance réelle de former 4 recombinaisons différentes qui se trouvent juste sur des sites de liaison pour le virus. Mais si cela ne vous convainc pas, il y a une caractéristique spéciale de l’insert 4 que nous devons examiner.
Nous revenons maintenant aux nucléotides, les G-A-C-T qui composent la séquence codant pour les acides aminés dont nous avons parlé jusqu’à présent. La séquence de référence originale du génome du coronavirus a pour ID dans la banque de données NC_045512.1 et peut être consultée à l’adresse suivante : https://www.ncbi.nlm.nih.gov/nuccore/NC_045512.1
Vous pouvez jouer avec la séquence du génome en utilisant BLASTn (pour nucléotide) en allant sur cette page et en sélectionnant « Run BLAST » dans la colonne de droite, ce qui vous amènera à une page BLAST similaire à celle des protéines ci-dessus.
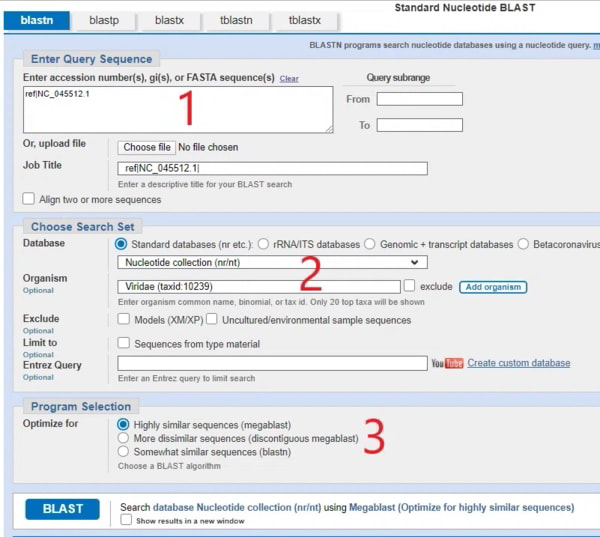
De la même manière, vous entrez NC_045512.1 (ou la mise à jour NC_045512.2) dans la première case, choisissez les options indiquées dans la partie marquée [2], sélectionnez megablast dans la partie 3 et cliquez sur go, et vous obtiendrez une liste des génomes SAR-CoV-2 qui correspondent (évidemment). Je ne montrerai pas cet écran car il n’est pas important ici, mais voici l’écran que vous obtenez si vous regardez deux séquences étroitement appariées et vous pouvez cliquer sur « CDS feature » pour superposer la séquence d’acides aminés. Vous vous retrouverez avec des pages qui ressemblent à ceci :
Dans cette section particulière, vous pouvez voir qu’il s’agit d’une séquence de la « protéine spike » (glycoprotéine de surface) et que les nucléotides sont étiquetés 23548…23771 (d’environ 30 000 nucléotides ou bases, c’est-à-dire G-C-A-T). [NB : en fait, il s’agit d’ARN et il devrait y avoir un U à la place de chaque T, mais BLAST le compense automatiquement pour des raisons de simplicité]. Le plus petit nombre est le nombre d’acides aminés dans la séquence de la protéine, donc pour chaque 3 nucléotides, le nombre augmente d’un acide aminé. Le point fort est l’acide aminé 677 (Q) à 686 (S), ce qui donne 677→686 = QTNSPRRARS.
Maintenant, c’est vraiment intéressant parce que non seulement nous avons vu que la section QTNS est dérivée du VIH, mais il y a quelque chose de très spécial au sujet du PRRAR adjacent parce que c’est un site de clivage de la furine et comme nous l’avons vu, ceux-ci n’existent pas dans ce type de virus semblable au SRAS. Il s’agit d’une insertion dans le génome viral, mais personne ne sait vraiment comment elle est arrivée là (tout comme les séquences du VIH). Pour savoir d’où elle vient, nous devons regarder au-delà de la séquence d’acides aminés et revenir à la séquence du génome.
La séquence génomique que vous pouvez voir pour cette séquence d’acides aminés est : CAGACTAATTCTCCTCGGCGGGCACGTAGT, soit 30 nucléotides codant pour 10 acides aminés. Pour que cette séquence soit le fruit du hasard, il faudrait un nombre infiniment petit. Elle doit donc être apparue quelque part (c’est-à-dire à partir d’un autre virus) ou bien une partie de cette séquence doit être synthétique. Nous allons donc l’analyser par BLAST(n), en excluant cette fois les « constructions synthétiques » de notre recherche (car nous recherchons des virus réels, pas des virus synthétiques). Qu’obtenons-nous ?
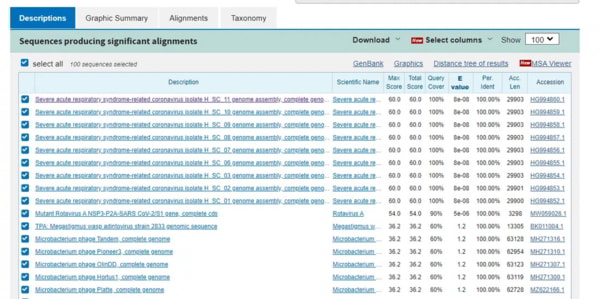
Maintenant que vous êtes habitués à ces affichages, nous pouvons constater que les seules séquences virales présentes ici sont synthétiques, et si vous cliquez sur chacune d’entre elles, vous trouverez leur date d’enregistrement après février 2020. En d’autres termes, aucun virus existant ne possède cette séquence génétique. C’est étrange, car pour qu’un virus puisse acquérir une grande séquence comme celle-ci, il doit l’obtenir d’un autre organisme. Il n’a pas de laboratoire pour manipuler les séquences génétiques, les chauves-souris non plus (d’où la petite blague de Jikky la souris de laboratoire)…
Il est assez facile de changer un seul nucléotide (une mutation ponctuelle ou SNP) ou même d’insérer ou de supprimer des nucléotides (ce qui est moins courant) mais d’insérer 20 ou 30 nucléotides avec un code qui fonctionne ? Non, cela doit provenir d’un autre virus ou bien cela a été fait en laboratoire.
Alors, d’où vient ce code ? Eh bien, il s’avère que BLAST peut nous dire – avec un certain degré de certitude – d’où vient une partie de ce code, en particulier la partie qui code pour la section PRRAR (le site de clivage de la furine qui est si unique).
La partie qui nous intéresse se trouve dans l’indice de Jikky. CTCCTCGGCGGGCACGTAG. Faisons un BLAST
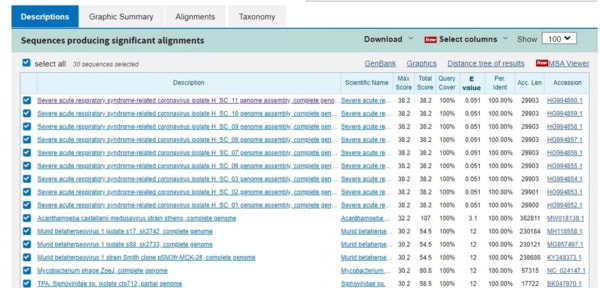
Ce que vous voyez, ce sont les mêmes séquences (SARS-CoV-2 mal classé) dans les 9 premiers résultats, puis aucun des résultats restants ne présente de correspondance 19/19. Cela signifie qu’aucun virus connu de l’homme ne possède cette séquence particulière dans son génome avant la découverte du SRAS-Cov-2. Alors, d’où vient-il ? Pour cela, vous devez choisir une autre base de données. Revenons à l’écran d’interrogation de BLASTn et changeons l’option de base de données pour « Patent sequences (pat) ». Supprimez toutes les exclusions et exécutez le BLAST.
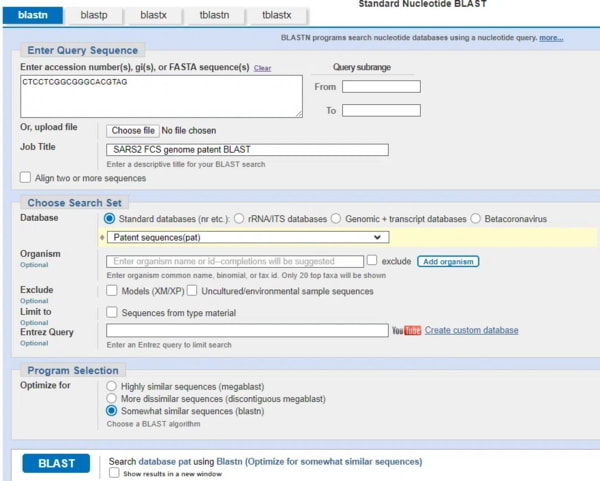
Les résultats doivent être passés au crible car le haut de la liste comprend des résultats de brevets de cette année. Ils sont préfixés WO2021 et WO2020 et peuvent donc être ignorés. Juste en dessous se trouvent les brevets qui nous intéressent. Je n’ai mis en évidence que les trois premiers, mais la liste entière comprend de nombreux brevets détenus par la même entreprise. Il suffit de cliquer sur le numéro d’accession à droite.
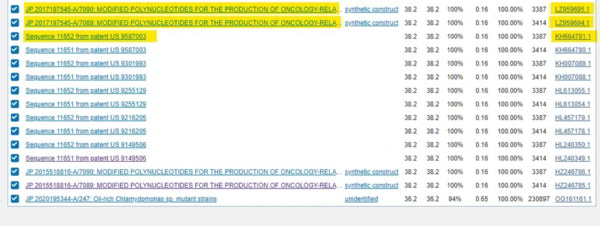
Alors faisons-le et voyons quelle société, que nous connaissons tous (maintenant), est une société pharmaceutique qui n’a jamais produit un médicament fonctionnel et qui a pourtant une capitalisation boursière de plus de 80 milliards de dollars…
Oui, c’est exact. Chacun de ces brevets qui contient cette séquence 19nt (dont la probabilité d’apparaître par hasard est inférieure à 1 sur un milliard) provient de Moderna. (Notez que la séquence est en fait la séquence complémentaire inverse, mais c’est probablement un résultat direct des lignées cellulaires dans lesquelles elle s’est produite – des lignées cellulaires MSH3_mutées conçues pour développer des vaccins contre le cancer, le brevet Moderna portait en fait sur un gène MSH3 muté à cette fin).
Pour que cette séquence soit apparue dans ce virus, il fallait que le virus fabriqué avec ses inserts VIH ait été infecté dans des lignées cellulaires brevetées fournies par Moderna, qui possédaient cette séquence unique qu’on ne trouve dans aucun autre virus.
En théorie, rien n’est impossible en science, en médecine ou en génomique. Un virus du SRAS émergeant naturellement avec 3 insertions de VIH sur ses sites de liaison et contenant également un site de clivage de la furine qui n’existe pas dans la nature mais qui existe dans un brevet Moderna… c’est vraiment n’importe quoi. Il n’existe pas. Un éléphant rose volant serait un million de fois plus probable.
- Lire aussi : Un expert en génétique lié à l’OMS explique comment le Covid-19 a pu être fabriqué en laboratoire
- Des scientifiques de Wuhan prévoyaient de transmettre des « protéines spike chimériques du Covid » dans les populations de chauves-souris à l’aide de « nanoparticules pénétrant la peau »
- L’armée américaine confirme que Fauci a menti – il a approuvé la recherche sur le gain de fonction














